In Vivo
Pharmacokinetics
Pharmacokinetics (PK) is the study of time course of drug’s or compound’s concentration in the body (what the body does to the drug or compound). The concentration profile of a compound or drug in the body is the result of the processes of Absorption (A), Distribution (D), Metabolism (M) and Elimination (E) or ADME. Most importantly the concentration of a compound in the body is linked to its efficacy or toxicity. Hence, characterizing the PK of a compound early on in discovery programs helps in evaluation of its drug-like properties.
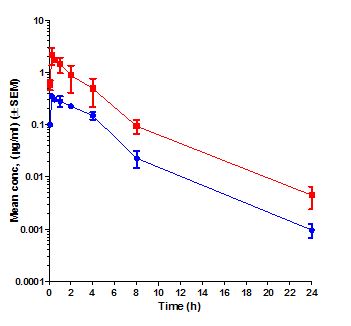
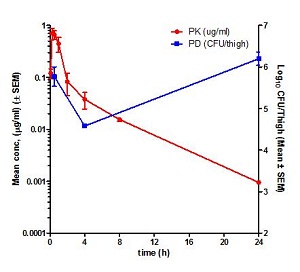
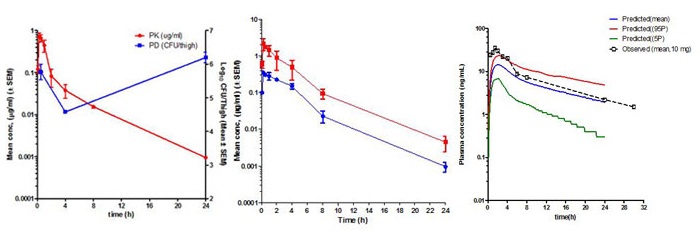
Fundamental PK parameters such as Volume of Distribution at steady state (Vss), Systemic Clearance, elimination half-life, the Area Under the Concentration Time curve in blood (AUC), Peak concentration in blood (Cmax), time to peak concentration in blood (Tmax), and Oral Bioavailability help in understanding the compound’s ability to show efficacy or toxicity in animals and human beings. Determination of PK parameters in preclinical studies help in optimizing molecules for efficacy and safety, progression to crucial pharmacology and toxicology studies, in prediction of human PK using Allometric scaling and Physiologically Based Pharmacokinetic (PBPK) and in Pharmacokinetic/Pharmacodynamic (PK/PD) modelling.