Proteolysis Targeting Chimeric Molecules: Tuning Molecular Strategies for a Clinically Sound Listening
The opportunity to drive the specific knockdown of the protein of interest (POI) through pharmacological entities called Proteolysis Targeting Chimeric molecules, or PROTACs, has become a possible therapeutic option with the involvement of these compounds in clinical trials for cancers and autoimmune diseases. The fulcrum of PROTACs pharmacodynamics is to favour the juxtaposition between E3 ligase activity and the POI, followed by the ubiquitination of the latter and its degradation by the proteasome system. In the face of an apparently modular design of these drugs, being constituted by an E3 ligase binding moiety and a POI-binding moiety connected by a linker, the final structure of an efficient PROTAC degradation enhancer often goes beyond the molecular descriptors known to influence the biological activity, specificity and PK, requiring a rational improvement through appropriate molecular strategies.
Paper from Sakamoto et al. in 2001 was the first original POC that cell degradation of specific protein of interest (POI), by ubiquitin-dependent proteolysis, can be artificially triggered by a properly conceived pharmacological agent displaying simultaneously a POI-ligand and a E3-ubiquitin–protein ligase moiety in the same molecule. Starting from the description of the basic principles underlying the activity of the PROTACs to the evaluation of strategies for the improvement of PK, PD and rational design, this review examines the elements that have been shown to be effective in allowing the evolution of these compounds from interesting POCs to potential aids of clinical interest.
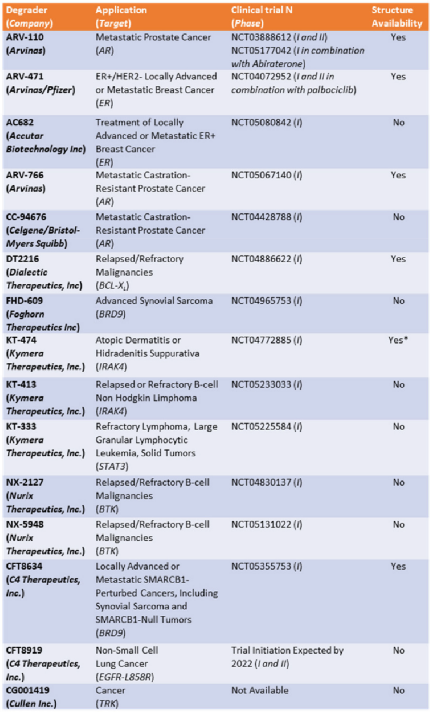
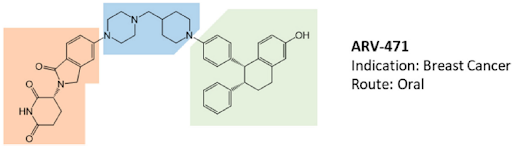
The opportunity to pharmacologically reprogram E3 ligase activity towards a specific POI was rapidly tapped to knock down disease-causing proteins but, differently from a genetic method, with properly conceived small-molecules. In the last two decades, there has been a real flourishing of new molecular approaches aimed at improving the specificity, efficacy, safety, and pharmacokinetic profile of this class of compounds. In 2019, the first PROTAC drug ARV-110 entered clinical trial for the treatment of prostate cancer, and only 3 years later, there are at least fifteen clinical trials aimed to evaluate the clinical efficacy of as many new PROTACs in the context of neoplastic and chronic-degenerative pathologies. Following picture aims to summarize which molecular approaches proved to be successful in developing a new protein degradation enhancer involved in a clinical trial.
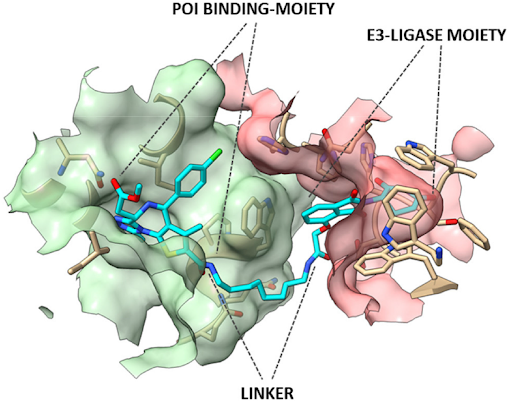
The fulcrum of the PROTAC’s pharmacodynamics is to allow the juxtaposition between E3 ligase and the POI through the formation of a stable ternary complex, within which the ubiquitination of the POI can take place. Ubiquitinated POI will then be delivered to the proteasome for degradation. The E3 ligase-mediated ubiquitination is actually the last in a multistep process involving the ATP-dependent activation of ubiquitin, operated by E1 enzymes, in the transfer of activated ubiquitin to a specific Cys-residue of one member of the ubiquitin-conjugating E2 enzyme family and the donation of conjugated-ubiquitin to protein substrates through the ubiquitin–protein ligase E3 complexes.
Among the 600 members of the E3 family, only few can be recruited according to the availability of recognized specific ligands. These are the ligands of cereblon (CRBN), such as thalidomide, pomalidomide and lenalidomide; ligands of the cellular inhibitor of apoptosis protein 1 (cIAP1), such as bestatin; ligands of the mouse double-minute 2 homolog (MDM2), such as nutlin and the ligands of Von Hippel-Lindau (VHL).
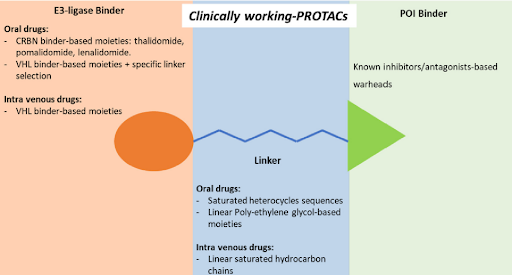
To date, of the fifteen compounds included in clinical trials, the only two recognized VHL-based degradation enhancers are ARV-766 (from Arvinas, CT, USA) and DT2216 (from Dialectic Therapeutics, TX, USA). In contrast, CRBN-based PROTACs appear as the first-choice option in making a compound of clinical relevance. This is essentially attributable to pharmacokinetic reasons since thalidomide and derivatives, such as E3 ligase-binding moiety, show lower molecular weight and favorable hydrogen bond donors/acceptors balance and CLogP. In this regard, working on available PK cassette studies on PROTACs and conventional small molecules in animal models showed that PROTACs derived from the prototypical parent ligands thalidomide, pomalidomide, and lenalidomide, display oral bioavailability greater than 30% compared with other homologous molecules.
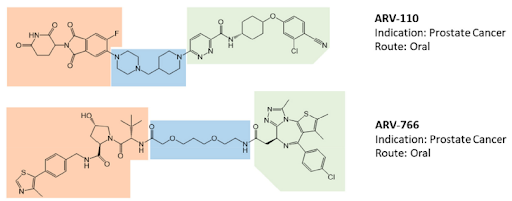
The VHL-based PROTAC, DT2216 requires IV administration, unlike the other compounds under evaluation. Interestingly, despite the fact that the molecule CFT8634 (from C4 Therapeutics, MA, USA) is claimed as a CRBN-based PROTAC, the molecule’s structure shows no thalidomide derivatives other than a residual glutarimide structure, probably representing the result of a deep rationalization in drug design, involving interaction with the E3 ligase.
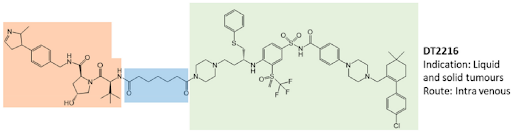
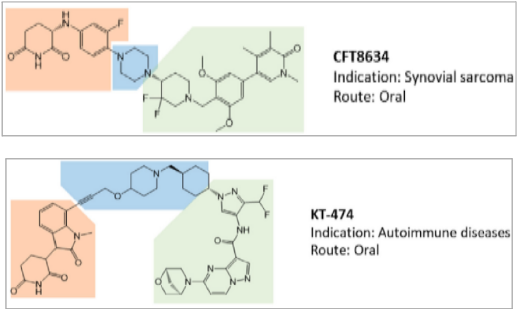
The molecular portions underlined in orange, green, and blue represent the E3 ligase-binding moiety, the protein of interest-binding moiety (POI) and the linker, respectively. These PROTACs act through a complex dynamic process, which implies the recognition by the unbound PROTAC at one side of the first protein (either the E3 ligase or the POI), the recognition by the bound PROTAC at the other side of the second protein (either the POI or the E3 ligase), and finally, the rearrangement of the supramolecular complex to the effective ternary complex. Accordingly, the PROTAC linker must not only possess the right length (so that the small molecule can interact with both proteins at the same time) but also proper flexibility. Indeed, during the last step, the PROTAC linker must collapse to allow the proteins to get in close contact. Based on these findings, PROTACs are likely to display PK/PD features of small molecules, thereby representing an evolution of PROTAC as the anticancer therapy.
- 1. Luca De Toni et al. Proteolysis Targeting Chimeric Molecules: Tuning Molecular Strategies for a Clinically Sound Listening. Int. J. Mol. Sci. 2022, 23, 6630-6643.
- 2. Sakamoto, K. M. et al. Protacs: Chimeric Molecules That Target Proteins to the Skp1-Cullin-F Box Complex for Ubiquitination and Degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 8554-8559.
- 3. Hughes, S.J. et al. The Rise and Rise of Protein Degradation: Opportunities and Challenges Ahead. Drug Disc. Today 2021, 26, 2889-2897.
- 4. Mullard, A. First Targeted Protein Degrader Hits the Clinic. Nat. Rev. Drug Disc. 06 March, 2019.
- 5. Békés, M. et al. PROTAC Targeted Protein Degraders: The Past Is Prologue. Nat. Rev. Drug Discov. 2022, 21, 181–200.