Optimization of mTOR Inhibitors Using Property-Based Drug Design and Free−Wilson Analysis for Improved In-Vivo Efficacy
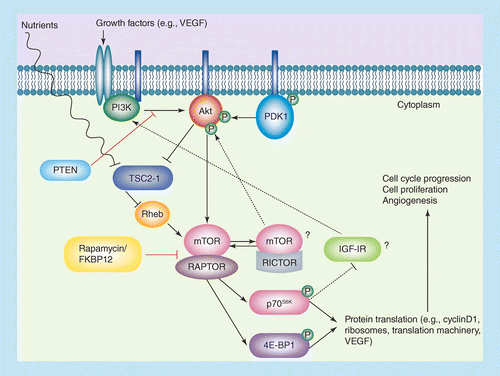
The mammalian target of rapamycin (mTOR) is a serine/threonine protein kinase and a member of (PI3K) phosphatidylinositol 3-kinase related kinase family with sequence similarity to PI3Ks. mTOR regulates cell growth, cell survival, cell proliferation, cell motility, protein synthesis, autophagy, and transcription. Due to its involvement in these processes, mTOR inhibitors have been sought for glycogen storage disease, anti-cancer, anti-aging, rheumatoid arthritis etc.
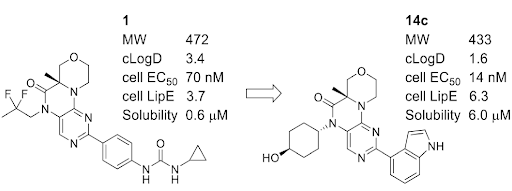
Using property-based drug design, a series of selective mTOR inhibitors based on the oxazinopteridine scaffold was optimized. Synthetic efforts resulted in compound 14c, that showed improved EC50, solubility and similar in-vivo efficacy as compared to the lead molecule 1 at 1/15 the dose, a result of its improved drug-like properties.
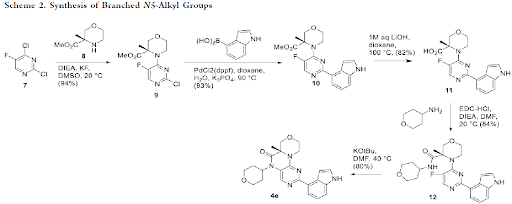
Synthesis of the representative compound 4e was achieved using the following synthetic scheme. After nucleophilic aromatic substitution in DMSO at rt, more active chloride of the dichloride derivative 7 gave morpholino pyrimidine intermediate 9. Suzuki reaction using PdCl2(dppf) and K3PO4 installed the desired 4-indolyl group to give compound 10 (93% yield). Hydrolysis of the ester proceeded smoothly using LiOH in dioxane to give acid derivative 11 in excellent yield. A standard amide coupling using EDC.HCl and Hunig’s base in DMF gave the desired intermediate 12 in 84% yield. Finally, the intramolecular ring closure proceeded in good yield at 40 °C once the amide was deprotonated with potassium tert-butoxide in DMF to furnish the desired oxazinopteridine derivative 4e.
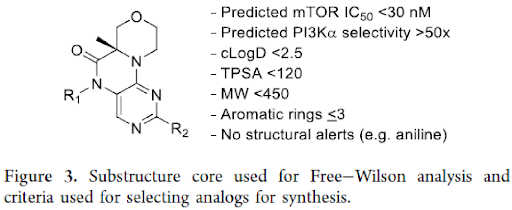
For the final round of designing, a Free−Wilson analysis was performed on the existing analogs to find new combinations. Free−Wilson analysis assumes additive SAR and suggested that the enzyme potencies for both mTOR and PI3Kα were indeed additive that allowed to predict not only potency but also selectivity. The Free−Wilson analysis included analogs with the substructure shown in Figure 3 and created all possible combinations of R1 and R2 analogs. In addition to predicting the enzyme potency and selectivity (ratio of predicted PI3Kα potency over predicted mTOR potency), physicochemical properties such as cLogD, tPSA and MW were also calculated.
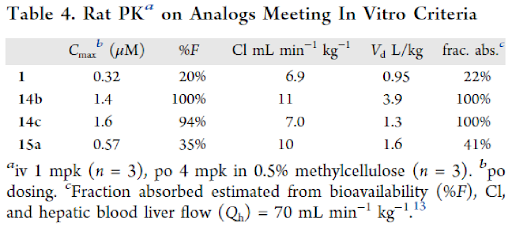
Based on the in-vitro potency, four compounds 1, 14b (IC50 = 14 nM), 14c (IC50 = 13 nM) and 15a (IC50 = 12 nM) were subjected to Rat PK studies to assess the exposure levels and the fraction absorbed (Table 4). Both analogs with the 4-indole and cyclohexanol groups (14b and 14c respectively) had excellent bioavailability, corresponding fraction absorbed as well as low clearance and high exposure. Both these analogs met criteria for the desired improvement over lead molecule 1. However, compound 15a showed lower bioavailability and had lower exposure levels.
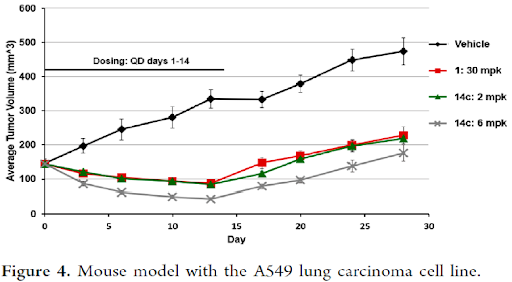
Based on the improved rat PK for compound 14c, it was dosed at 2 and 6 mpk along with compound 1 at 30 mpk to achieve similar or better in-vivo exposure profile in a mouse xenograft model with the lung carcinoma cell line A549 (Figure 4). All dosing groups showed less than 20% body weight loss throughout the duration of the study. Both 30 mpk dose of 1 and 2 mpk dose of 14c fully inhibited tumor growth, with modest tumor burden reduction observed while dosing for 14 days. The 6 mpk dose of 14c reduced the tumor volume by more than 50%. Analog 14c showed comparable in-vivo efficacy as compound 1 but at 1/15 the dose, which is likely a combination of the higher cellular potency (14 nM vs 70 nM) and improved oral absorption (fraction absorbed 100% vs 22%). The improved absorption is consistent with 10-fold increase in thermodynamic solubility at pH 6.8 (6.0 μM for 14c vs 0.6 μM for 1).
- 1. Sean T. Murphy et al. Optimization of mTOR Inhibitors Using Property-Based Drug Design and Free−Wilson Analysis for Improved In-Vivo Efficacy. ACS Med. Chem. Lett, ASAP article.
- 2. Chen, Y. et al. Research Progress of mTOR Inhibitors. Eur. J. Med. Chem. 2020, 208, 112820.
- 3. Bonazzi, S. et al. Discovery of a Brain-Penetrant ATP-Competitive Inhibitor of the Mechanistic Target of Rapamycin (mTOR) for CNS Disorders. J. Med. Chem. 2020, 63 (3), 1068−1083.
- 4. Charles J. Malemud. The PI3K/AKT/PTEN/mTOR pathway: A Fruitful Target for inducing Cell Death in Rheumatoid Arthritis. Future Med. Chem. 2015, Vol. 7, No. 9, 1137-1147.
- Author : Dr. Sachin Madan-Director – Medicinal Chemistry at TheraIndx Lifesciences pvt ltd