Discovery of Orally Available and Brain Penetrant AEP Inhibitors
Alzheimer’s Disease (AD) is the most widespread form of dementia, with one of the pathological hallmarks being the formation of neurofibrillary tangles (NFTs). These tangles consist of phosphorylated Tau fragments. Asparagine endopeptidase (AEP) is a key Tau cleaving enzyme that generates aggregation prone Tau fragments. Inhibition of AEP to reduce the level of toxic Tau fragment formation could represent a promising therapeutic strategy. The first orthosteric, selective, orally bioavailable, and brain penetrant inhibitors are reported with an irreversible binding mode. The development of the series starting from reversible molecules and the link between inhibition of AEP and reduction of Tau N368 fragment both in vitro and in vivo is also highlighted.
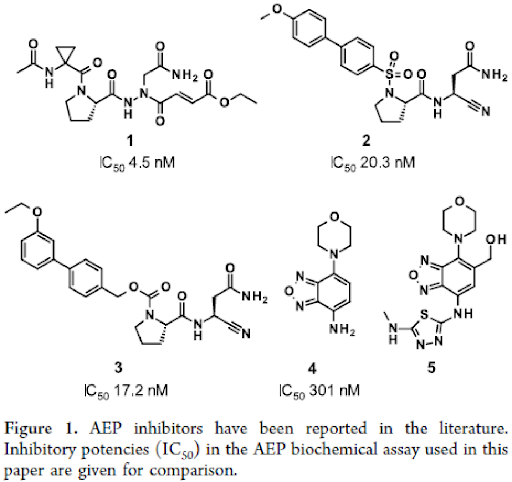
Efforts to find AEP inhibitors (compounds 1-3) were motivated by the role of AEP in antigen processing and as potential target for cancer therapy, based on the observation of its overexpression in various carcinoma as well as the link to poor prognosis and metastases formation. In addition to this, compounds 4 and 5 were also reported to exhibit pro-cognitive effects in vivo, presumably acting via an allosteric mechanism of inhibition. However, these inhibitors suffered from poor permeability due to high polarity as well as high protein binding.
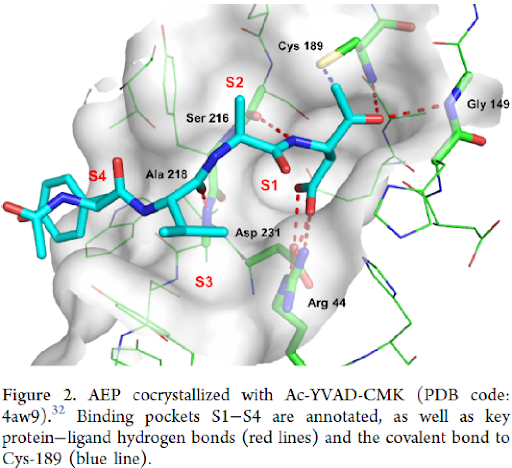
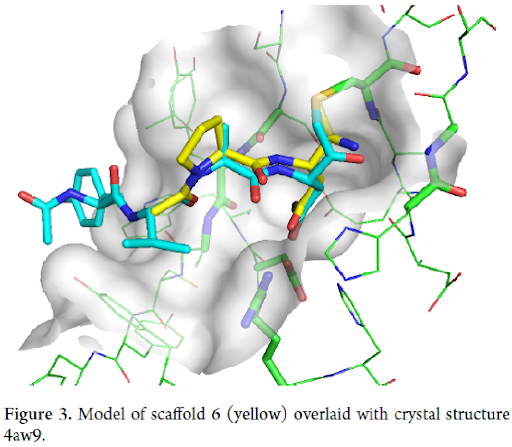
As a starting point, nitriles of general formula 6 were chosen, in which the cyano group should bind covalently to Cys-189, the propanamide substituent mimic P1 Asn residue and the secondary amide NH and the tertiary amide C=O interact with backbone of Ser-216 and Ala-218, respectively (Figures 2 and 3).
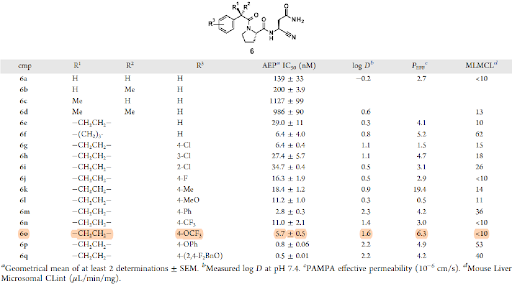
First SAR studies (Table 1) revealed that aryl acetamides gave best potency, which could be increased further by a factor of 5−20 by installing substituents such as cyclopropyl or cyclobutyl in the α position (compounds 6e & 6f vs 6a). Additionally, lipophilic substituents in the para position of the aryl ring had beneficial effects on both potency and permeability. A balance between lipophilicity, high permeability, and low clearance was crucial to get compounds with good in vivo (PK) profile. Likewise, we identified compound 6o as the first tool that was further profiled. Besides an excellent selectivity demonstrated in both a 10 protease and 50 off-target (CEREP) panel, compound 6o showed a decent PK profile in mice with an oral bioavailability of 41% and brain penetration with a Kp,uu of 0.22.
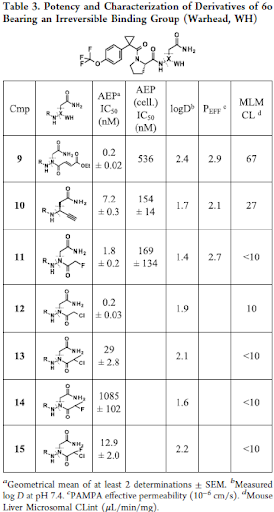
Next, a more sustained, potentially irreversible inhibition was envisioned to ensure full blockage of AEP. Therefore, the warhead was varied, and various irreversible reactive groups were attached to the corresponding hydrazide scaffold (Table 3). Enzymatic inhibitory activity after 0.5 h compound incubation overall correlated with reactivity at the electrophilic center, and chloro acetamide 12 was identified as the most potent one but had stability issues in the profiling assays. Gradually decreasing the reactivity led to the identification of fluoroacetamide 11 that combined good potency in secondary functional assays with a decent selectivity, DMPK profile and was further characterized in vivo. Also, compound 11 showed moderate bioavailability, a decent half-life in mice after p.o. administration, while the brain penetration was poor, driven by low passive cellular permeability.
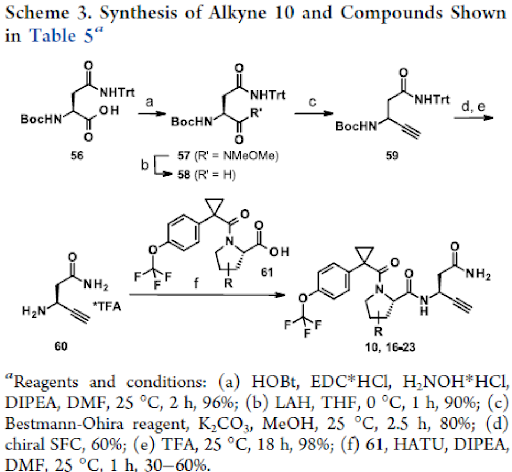
Next, we turned our attention to alkyne 10 to improve the brain penetrance as well as metabolic stability. Alkynes have been described as irreversible warheads and form a thioenol ether with cysteines, if positioned in close proximity in the binding pocket. They are more lipophilic than fluoro acetamides and thus deemed more appropriate for achieving brain penetration. Further, metabolite identification studies with compound 6o revealed the proline part to be a metabolic hot spot. Therefore, a variety of proline replacements in compound 10 were explored next and their influence on potency and metabolic stability was investigated (Table 5).
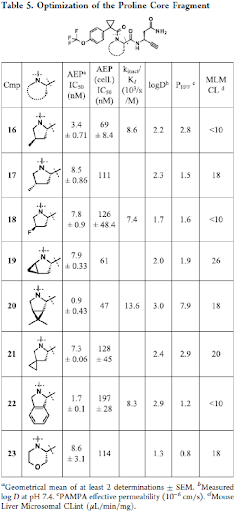
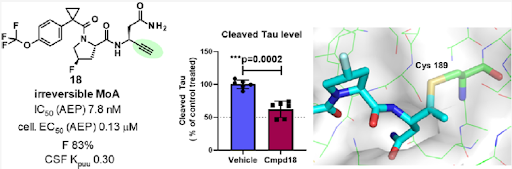
Overall, considering the further characterization, e.g., stability against hepatocytes, P-gp transporter efflux ratio and permeability, compound 18 showed the most balanced profile. Finally, it was confirmed with compound 18 that the formation of the fragment is also suppressed in vivo. Indeed, after 5d treatment in Tau transgenic mice expressing the 2N4R isoform of Tau containing the P301L mutation, compound 18 showed significantly inhibited activity of AEP in brain and complete inhibition in all peripheral organs tested. At the same time, reduced formation of the Tau N368 fragment was observed thereby, confirming the result measured in vitro although the effect on total Tau is not significant.
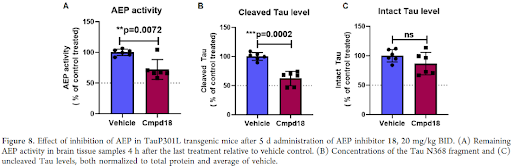
The basic hypothesis that a reduced amount of Tau N368 fragment will lead to reduced Tau aggregation and ultimately to improved cognition remains to be shown in a long-term experiment. Synthesis of the alkyne compound 10 and other derivatives as shown in Table 5 is being highlighted below.
- 1. Bartels, Bjorn et al. Discovery of Orally Available and Brain Penetrant AEP Inhibitors. J. Med. Chem. 2023, 66 (24), 17026-17043.
- 2. Vaquer-Alicea, J. et al. Tau Strains Shape Disease. Acta Neuropathol. 2021, 142 (1), 57−71.
- 3. Bogyo, M. et al. Synthesis and Evaluation of Aza-Peptidyl Inhibitors of the Lysosomal Asparaginyl Endopeptidase, Legumain. Bioorg. Med. Chem. Lett. 2012, 22 (3), 1340−1343.
- 4. Williams, R. et al. Identification and SAR Exploration of a Novel Series of Legumain Inhibitors. Bioorg. Med. Chem. Lett. 2019, 29 (12), 1546−1548.
- 5. Alquezar, C. et al. Tau Post-Translational Modifications: Dynamic Transformers of Tau Function, Degradation and Aggregation. Front. Neurol. 2021, 11, No. 595532.