Discovery of Futibatinib: The First Covalent FGFR Kinase Inhibitor in Clinical Use
Deregulating fibroblast growth factor receptor (FGFR) signalling is a promising strategy for cancer therapy. The discovery of compound 5 (TAS-120, futibatinib), a potent and selective covalent inhibitor of FGFR, starting from a unique dual inhibitor of mutant epidermal growth factor receptor (EGFR) and FGFR (compound 1), is reported. Compound 5 inhibited all four families of FGFRs in the single-digit nM range and showed high selectivity for over 387 kinases. Binding site analysis revealed that compound 5 covalently bound to the cysteine 491, highly flexible glycine-rich loop region of the FGFR2 ATP pocket. Futibatinib is currently in clinical trials for patients with oncogenically driven FGFR genomic aberrations.
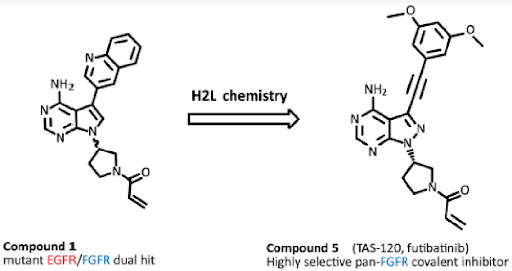
During systematic kinome screening of an in-house compound collection, pyrrolopyrimidine analogue 1 was identified as a unique hit with inhibitory activities of both FGFR2 and EGFRdel19/T790M mutations, with a half maximal inhibitory concentration (IC50) of 57 and 1.2 nM, respectively. Because acrylamide moiety has been widely used as an electrophile that can bind to thiol group of the cysteine residues in covalent drug discovery, this compound was expected to bind covalently to both the kinases. Compound 1 was considered good starting point to synthesize novel covalent inhibitors for both mutant EGFR and FGFR.
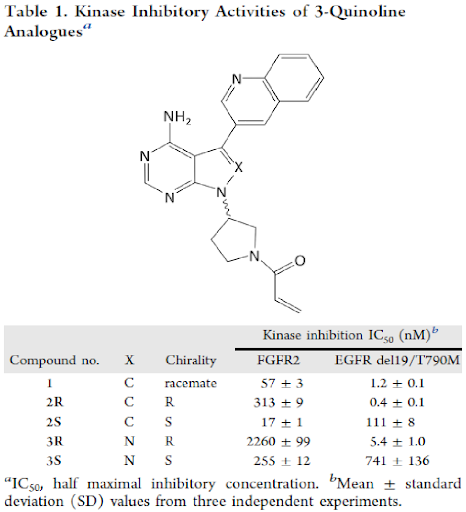
The enantiomers of compound 2 (pyrrolopyrimidine) and its analogues, pyrazolopyrimidine 3, were prepared to investigate the effect of stereochemistry of the pyrrolidine ring and the nitrogen atom at the X position on kinase inhibitory activities. All prepared compounds were tested using two kinase enzyme assays and the results are shown in Table 1. Each enantiomer exhibited a different trend of kinase inhibitory activity. EGFR or FGFR inhibitory activities of compounds were controlled solely by the stereochemistry at the 3-position of pyrrolidine ring. The R-pyrrolidine analogues 2R and 3R showed more potent inhibitory activity against mutant EGFR than FGFR2. However, the S-pyrrolidine analogues 2S and 3S showed better FGFR2 inhibition. Although 2S showed approximately 15-fold more potent inhibitory activity on FGFR2 than 3S, 3S was selected as an initial lead molecule for rapid SAR generation, considering the synthetic ease of the pyrazolopyrimidine core. This core was chemically examined in the development of ibrutinib and had a better nucleophilic reactivity at 1-position of nitrogen.
First, Suzuki, Sonogashira and amide coupling reactions were used to introduce 3,5-dimethoxybenzene moiety and the different types of bonds affected the inhibitory activity on FGFR2, as shown in Table 2. For example, the activity of direct-bonded compound 4 obtained from Suzuki coupling was almost the same as that of quinoline compound 3S, probably owing to insufficient occupation of the target space. However, the alkyne derivative 5 having an extended straight molecule, showed an approximately 200-fold increase for FGFR2 kinase inhibitory activity over the 3-quinolinyl derivative 3S.
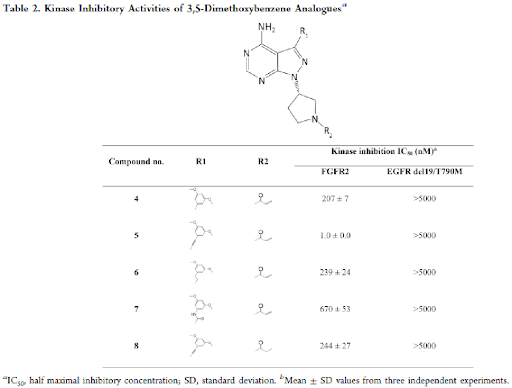
Next, the hydrogenated analogue 6 showed decreased FGFR2 inhibitory activity (240-fold) versus compound 5. Similarly, the activity of the amide analogue 7 was 670-fold lower than that of compound 5. As an initial SAR study, only alkyne compound 5 achieved a single-digit nM inhibitory activity on FGFR2. These data suggest that the straight-chain alkyne could correctly place the 3,5-dimethoxy benzene ring in the well-known unique hydrophobic pocket of FGFR. Propionamide analogue 8, without covalent binding ability, was prepared to determine the effect of the acrylamide warhead moiety on FGFR2 inhibition. Expectedly, compound 8 reduced the inhibitory activity (240-fold) as compared to compound 5, indirectly confirming the covalent inhibitory mechanism. Also, compound 5 (TAS-120, futibatinib) was evaluated for inhibitory activity against FGFR1, FGFR3 and FGFR4 and showed IC50 values of 1.8, 1.6, and 3.7 nM, respectively. It showed good microsomal stability profile in both human and mouse (66 and 69% remaining respectively).
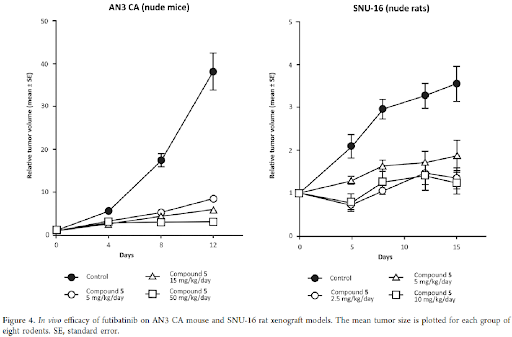
The in vivo antitumor effect of futibatinib was tested in two xenograft models implanted with AN3 CA human endometrial carcinoma cells in nude mice and SNU-16 gastric carcinoma cells in nude rats. The AN3 CA cell line expresses FGFR2 with K310R and N549K point mutations; the SNU-16 cell line exhibits FGFR2 amplification. The doses used in efficacy studies were based on FGFR inhibitory activity, as determined by pharmacodynamic analysis. Significant antitumor efficacy was confirmed at the lowest dose in both models, consistent with results from the pharmacodynamic study as shown in Figure 4. No signs of toxicity were observed for any dose used in these studies.
- 1. Satoru Ito et al. Discovery of Futibatinib: The First Covalent FGFR Kinase Inhibitor in Clinical Use. ACS Med. Chem. Lett, 2023, 14, 396-404.
- 2. Goyal, L. et al. Futibatinib, an Irreversible FGFR1−4 Inhibitor, in Patients with Advanced Solid Tumors Harboring FGF/FGFR Aberrations: A Phase I Dose-Expansion Study. Cancer Discov. 2022, 12, 402−415.
- 3. Lu, X. et al. Discovery of Cysteine-Targeting Covalent Protein Kinase Inhibitors. J. Med. Chem. 2022, 65, 58−83.
- 4. Helsten, T. et al. The FGFR Landscape in Cancer: Analysis of 4853 Tumors by Next Generation Sequencing. Clin. Cancer Res. 2016, 22, 259−267.
- Author : Dr. Sachin Madan- Director Medicinal Chemistry at TheraIndx Lifesciences Pvt Ltd